Kevin Waters An Academic Portfolio
Solvating Structures
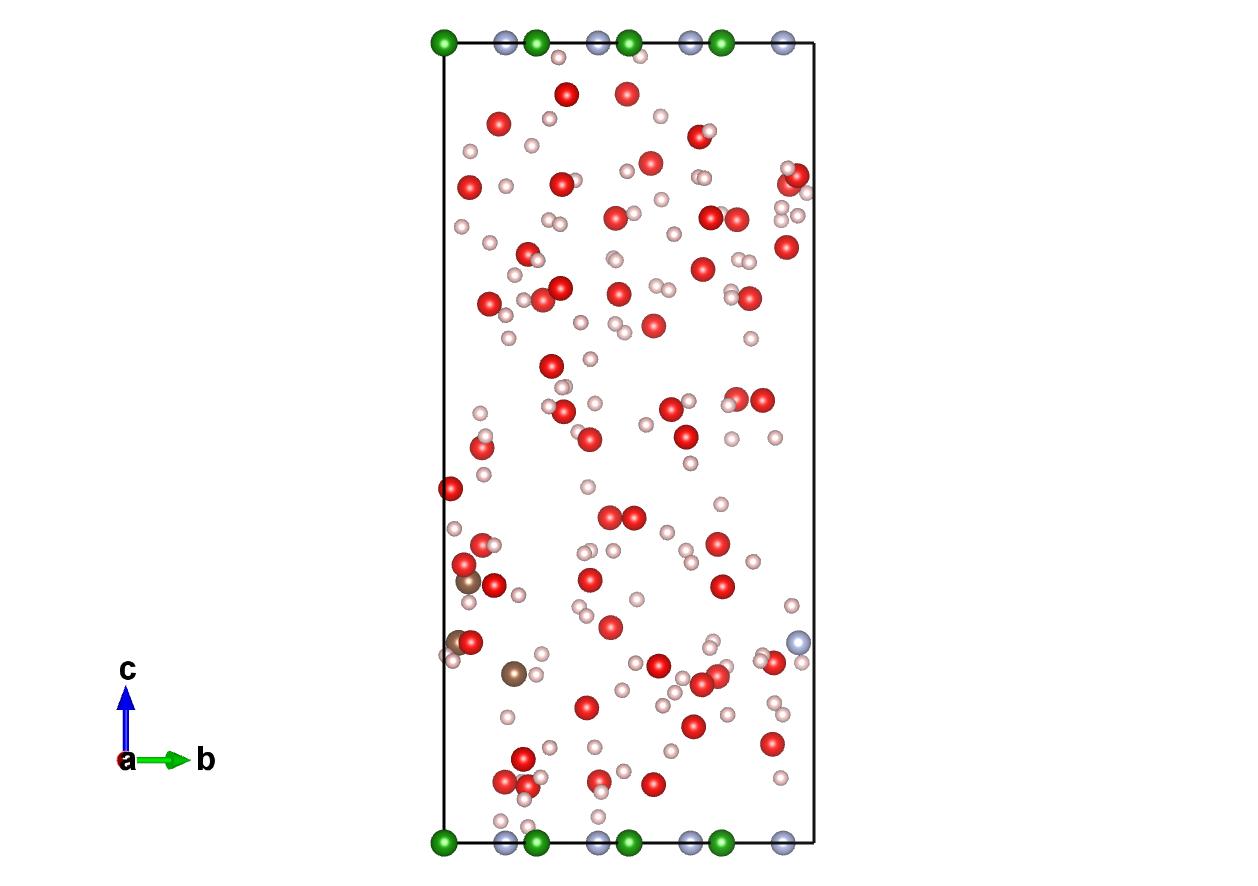
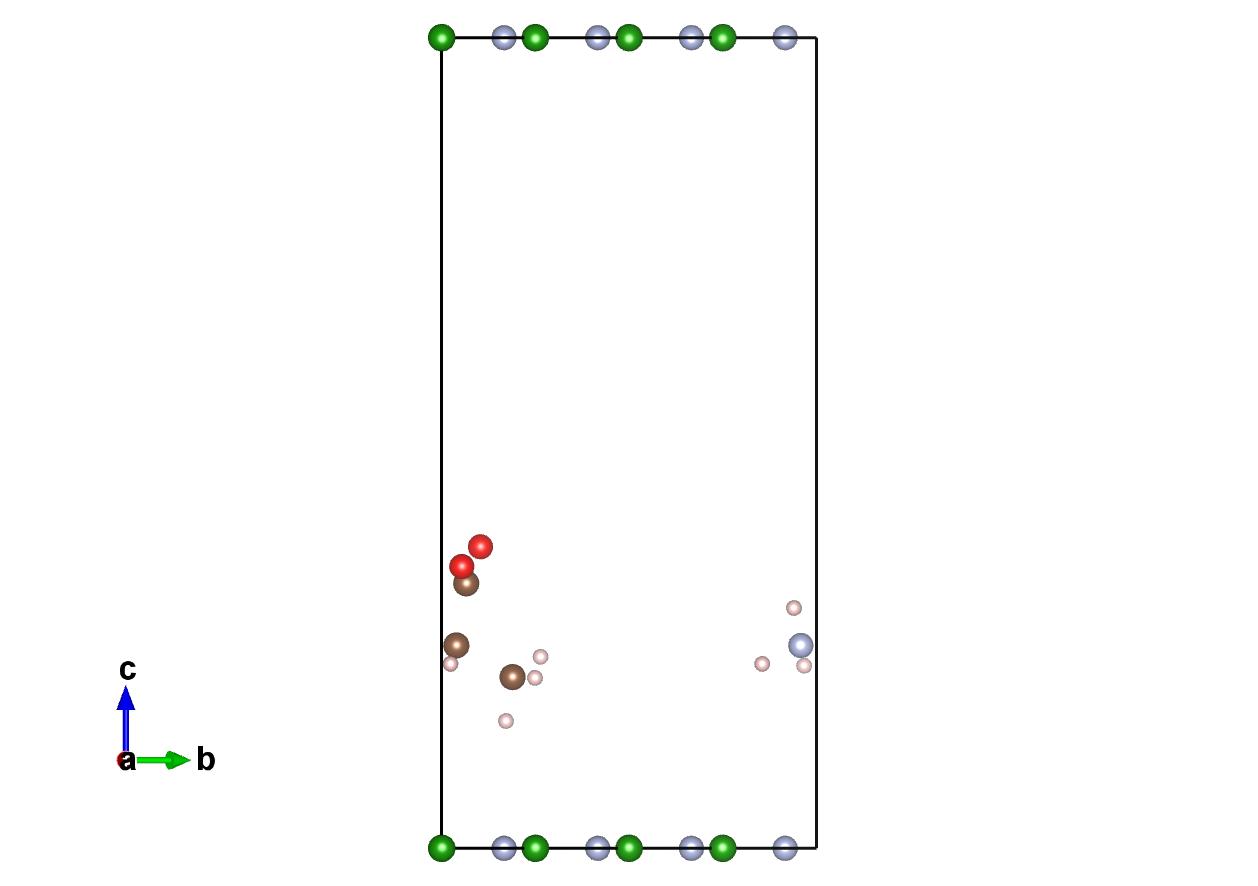
Here is a program that I am using to generate solvated structures for ab initio molecular dynamics simulations I am currently running in the software suite NWChem. It uses a Monte Carlo scheme to solvate a periodic structure. Random points in the cell are selected and a oxygen atom is placed there and if any atoms overlap with the van der Waal’s radius of water it is removed and the next site is sampled. The volume that is filled is calculated by taking the overall cell volume and subtracting the volume taken by the atoms already in the cell, again using the van der Waal’s radius. The algorithm runs quickly and usually takes less than a second to run.
The structure that is used is the structure class used by the Pymatgen library. The output is a structure object with the water molecules appended to the end of the atoms list. At this point you can take the structure and the library’s IO capabilities to generate the file format you desire.
Before
This is a unit cell with a monolayer of hexagonal boron-nitride with the Alanine amino acids place on top of the surface.
 The .cif file can be downloaded here.
The .cif file can be downloaded here.
Code
import numpy as np
from pymatgen.core.periodic_table import Element
base_density = 1.0 / 1E8**3 * 6.022E23 / 18.09 # H20/A**3
vdw_h2o = 1.4 # van der Waals radius in (Angstrom)
b = 0.96 # O-H bond lengths
omega = np.radians(104.5) # H-O-H bond angle (degrees)
def water(structure):
atom_index = len(structure.sites)
water_mol = 0
samples = 0
substrate_volume = 0
# Estimate volume of atoms that are not water
for site in structure.sites:
substrate_volume += 4 * np.pi * (site.specie.atomic_radius_calculated)**3
# Number of water molecules
water_approx = (structure.volume - substrate_volume) * base_density
# Populate water molecules
while water_mol < int(water_approx):
# Copy structure to a temporary structure
temp_structure = structure.copy()
# Generate point in the unit cell to place oxygen atom
x = rand.random() * structure.lattice.a
y = rand.random() * structure.lattice.b
z = rand.random() * structure.lattice.c
# Random angle for placement of first hydrogen atom
phi = rand.random() * 2 * np.pi
theta = rand.random() * np.pi
# First Hydrogen Position
xh1 = x + b * np.sin(theta) * np.cos(phi)
yh1 = y + b * np.sin(theta) * np.sin(phi)
zh1 = z + b * np.cos(theta)
# Second Hydrogen Position
sign = rand.randint(0,1)
xh2 = x + b * np.sin(theta - (-1)**(sign) * omega) * np.cos(phi)
yh2 = y + b * np.sin(theta - (-1)**(sign) * omega) * np.sin(phi)
zh2 = z + b * np.cos(theta - (-1)**(sign) * omega)
o1 = np.array([x, y, z])
h1 = np.array([xh1, yh1, zh1])
h2 = np.array([xh2, yh2, zh2])
temp_structure.append(Element("O"), o1, coords_are_cartesian = True)
h2o_valid = True
# Check all distances in the temp_structure to check if it fits
for i in range(0,len(temp_structure.sites) -1):
distance = temp_structure.get_distance(i,-1)
if distance <= vdw_h2o:
h2o_valid = False
break
# If molecule position is valid, append to the structure
if h2o_valid == True:
structure.append(Element("O"), o1, coords_are_cartesian = True)
structure.append(Element("H"), h1, coords_are_cartesian = True)
structure.append(Element("H"), h2, coords_are_cartesian = True)
water_mol += 1
samples += 1
print("Sampled Spaces : {}".format(samples))
print("Placed : {} \t Estimate : {}".format(water_mol, int(water_approx)))
return structure
After
The .cif file can be downloaded here.