Kevin Waters An Academic Portfolio
Projected Band Structure and DOS
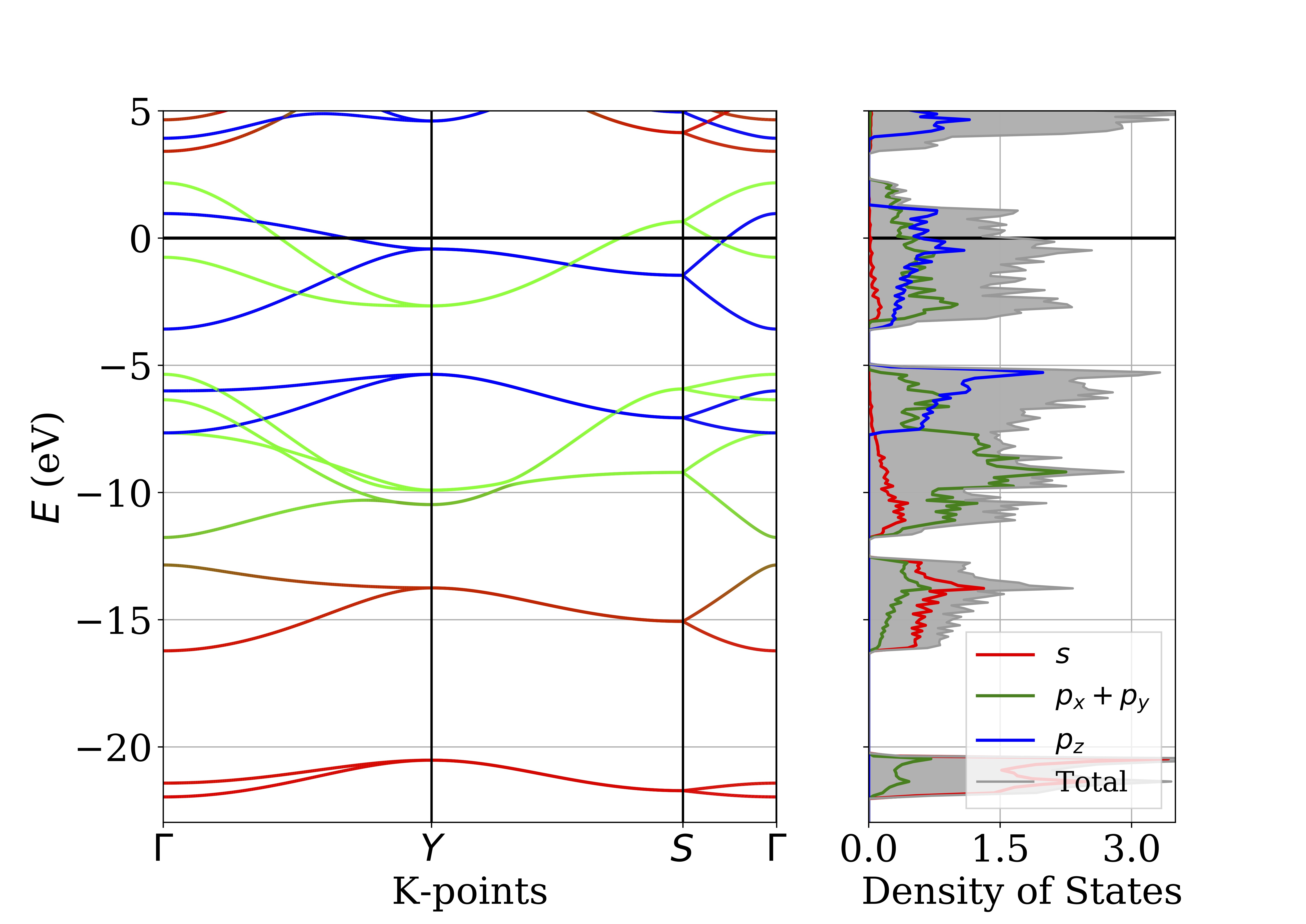
The program is based of the original posted by here. This is a code I still want to come back to and reorganize, but it is functional in its current state. It plots the orbital projected band structure and density of states for a VASP calculation. At the bottom of the page is a .zip file with the contents needed to give it a test run. Some manual inputs for kpoint spacing and labels (Can be fixed), found under the comment #labels.
An atom projected code will be coming soon.
Files Needed
1) Band structure vasprun.xml
2) Band structure KPOINTS
3) Band structure PROCAR
4) PDOS vasprun.xml
Code
from numpy import array as npa
import numpy as np
import math
import pymatgen
import sys
from pymatgen.io.vasp.outputs import Procar, Vasprun
from pymatgen import Structure
from pymatgen.electronic_structure.core import Spin, Orbital
import matplotlib.pyplot as plt
from matplotlib.collections import LineCollection
from matplotlib.gridspec import GridSpec
def rgbline(ax, k, e, red, green, blue, KPOINTS, alpha=1.):
#creation of segments based on
#http://nbviewer.ipython.org/urls/raw.github.com/dpsanders/matplotlib-examples/master/colorline.ipynb
pts = np.array([KPOINTS, e]).T.reshape(-1, 1, 2)
seg = np.concatenate([pts[:-1], pts[1:]], axis=1)
nseg = len(KPOINTS) -1
r = [0.5*(red[i]+red[i+1]) for i in range(nseg)]
g = [0.5*(green[i]+green[i+1]) for i in range(nseg)]
b = [0.5*(blue[i]+blue[i+1]) for i in range(nseg)]
a = np.ones(nseg, np.float)*alpha
lc = LineCollection(seg, colors=zip(r,g,b,a), linewidth = 2)
ax.add_collection(lc)
if __name__ == "__main__":
# Load Structure
structure = Structure.from_file("./BAND/POSCAR")
# Load Band Structure Calculations
bands_K = Vasprun("./BAND/vasprun.xml")
# Load Band Structure Calculations
bands = Vasprun("./BAND/vasprun.xml").get_band_structure("./BAND/KPOINTS", line_mode = True)
# projected bands
data = Procar("./BAND/PROCAR").data
# density of states
dosrun = Vasprun("./PDOS/vasprun.xml")
# labels (MANUAL INPUT NEEDED CURRENTLY HERE)
labels=[u"$\\Gamma$", u"$Y$", u"$S$", u"$\\Gamma$"]
# Number of points between kpoints, found in the KPOINTS file
step = 150
# general options for plot
font = {'family': 'serif', 'size': 24}
plt.rc('font', **font)
# set up 2 graph with aspec ratio 2/1
# plot 1: bands diagram
# plot 2: Density of States
gs = GridSpec(1, 2, width_ratios=[2,1])
fig = plt.figure(figsize=(11.69, 8.27))
ax1 = plt.subplot(gs[0])
ax2 = plt.subplot(gs[1]) #, sharey=ax1)
# set ylim for the plot
# ---------------------
emin = 1e100
emax = -1e100
# Set both fermi levels equal to the band fermi level
bands.efermi = dosrun.efermi #= 0
for spin in bands.bands.keys():
for b in range(bands.nb_bands):
emin = min(emin, min(bands.bands[spin][b]))
emax = max(emax, max(bands.bands[spin][b]))
emin -= bands.efermi + 1
emax -= bands.efermi - 1
# emin = -20
emax = 5
ax1.set_ylim(emin, emax)
ax2.set_ylim(emin, emax)
# makes an empty list with the length of the number of bands and kpoints
contrib = np.zeros((bands.nb_bands, len(bands.kpoints), 3))
# sum up atomic contributions and normalize contributions
# Sum over all bands
for b in range(bands.nb_bands):
# Sum over all K-Points
for k in range(len(bands.kpoints)):
s = 0
py_px = 0
pz = 0
# Sum over all atoms
for i in range(len(bands.structure.species)):
# Sum over the orbitals
# Select which atom type they belong to
# 0:s 1:py 2:pz 3:px 4:dxy 5:dyz 6:dz2 7:dxz 8:dx2_y2
for j in range(0,8,1):
if j == 0:
s += data[Spin.up][k][b][i][j]**2
if j == 1 or j == 3:
py_px += data[Spin.up][k][b][i][j]**2
if j == 2:
pz += data[Spin.up][k][b][i][j]**2
tot = s + py_px + pz
if tot != 0.0:
contrib[b, k, 0] = s / tot
contrib[b, k, 1] = py_px / tot
contrib[b, k, 2] = pz / tot
reciprocal = bands.lattice_rec.matrix/(2*math.pi)
# Empty lists used for caculating the distances between K-Points
KPOINTS = [0.0]
DIST = 0.0
# Create list with distances between Kpoints (Individual), corrects the spacing
for k in range(len(bands.kpoints)-1 ):
Dist = np.subtract(bands.kpoints[k+1].frac_coords,bands.kpoints[k].frac_coords)
DIST += np.linalg.norm(np.dot(reciprocal,Dist))
KPOINTS.append(DIST)
# plot bands using rgb mapping
for b in range(bands.nb_bands):
rgbline(ax1,
range(len(bands.kpoints)),
[e - bands.efermi for e in bands.bands[Spin.up][b]],
contrib[b,:,0],
contrib[b,:,1],
contrib[b,:,2],
KPOINTS)
# style
ax1.set_xlabel("K-points")
ax1.set_ylabel(r"$E$ (eV)")
ax1.grid()
# fermi level line at 0
ax1.hlines(y=0, xmin=0, xmax=len(bands.kpoints), color="k", lw=2)
TICKS = [0.0]
for i in range(step,len(KPOINTS)+step,step):
ax1.vlines(KPOINTS[i-1], emin, emax, "k")
TICKS.append(KPOINTS[i-1])
ax1.set_xticks(TICKS)
ax1.set_xticklabels(labels)
ax1.set_xlim(0, KPOINTS[-1])
# Density of states
# -----------------
ax2.set_yticklabels([])
ax2.grid()
ax2.set_xticks(np.arange(0, 3.5, 1.5))
ax2.set_xlim(0,3.5)
ax2.hlines(y=0, xmin=0, xmax=3.5, color="k", lw=2)
ax2.set_xlabel("Density of States")
# atom contribution
# Zero NP arrays
s_dos = np.zeros(len(dosrun.pdos[0][Orbital.s][Spin.up]))
py_px_dos = np.zeros(len(dosrun.pdos[0][Orbital.s][Spin.up]))
pz_dos = np.zeros(len(dosrun.pdos[0][Orbital.s][Spin.up]))
# Sum over atoms
for i in range(len(bands.structure.species)):
# Print Progress Labels
print("Summing Densities of State")
print(str(bands.structure.species[i]) + " (" + str(i + 1) + " of " + str(len(bands.structure.species)) + ")")
s_dos += npa(dosrun.pdos[i][Orbital.s][Spin.up])
py_px_dos += npa(dosrun.pdos[i][Orbital.px][Spin.up]) + \
npa(dosrun.pdos[i][Orbital.py][Spin.up])
pz_dos += npa(dosrun.pdos[i][Orbital.pz][Spin.up])
ax2.plot(s_dos,dosrun.tdos.energies - dosrun.efermi, \
"r-", label = "$s$", linewidth = 2)
ax2.plot(py_px_dos,dosrun.tdos.energies - dosrun.efermi, \
"g-", label = "$p_x + p_y$", linewidth = 2)
ax2.plot(pz_dos,dosrun.tdos.energies - dosrun.efermi, \
"b-", label = "$p_z$", linewidth = 2)
ax2.fill_between(dosrun.tdos.densities[Spin.up],
0,
dosrun.tdos.energies - dosrun.efermi,
color = (0.7, 0.7, 0.7),
facecolor = (0.7, 0.7, 0.7))
ax2.plot(dosrun.tdos.densities[Spin.up],
dosrun.tdos.energies - dosrun.efermi,
color = (0.6, 0.6, 0.6),
label = "Total")
ax2.legend(fancybox=False, shadow=False, prop={'size': 18},loc=4)
# Plotting
# -----------------
plt.savefig(sys.argv[0].strip(".py") + ".pdf", format="pdf")
File Download
The files can be downloaded here.
Written on March 18th, 2018 by Kevin WatersFeel free to share!